
En algún momento de nuestra vida, todos hemos sentido vértigo al contemplar la inmensidad del universo y comparar nuestra pequeña existencia dentro de él. Parte de este vértigo viene de nuestra incapacidad para comprender dimensiones tan inmensas, donde las cifras superan nuestra imaginación y nos hacen sentir insignificantes. Bajemos la vista por un momento, hacia el cajón de las medicinas de nuestra casa. Quizá os sorprenda saber que existe un pequeño universo en cada fármaco que tomamos. El denominador común de estos universos son los átomos. Ese comprimido que te salva de una mala jaqueca tiene más de 1020 átomos, organizados en forma de moléculas de paracetamol. Un uno seguido de veinte ceros, un número difícil de digerir.
El tipo, número y combinación de estos átomos es lo que determina las propiedades de todo lo que nos rodea, incluidos los fármacos. Trece átomos de carbono junto a dieciocho hidrógenos y dos oxígenos dan lugar al ibuprofeno. La combinación de ocho carbonos, nueve hidrógenos, dos oxígenos y, en este caso, un nitrógeno, conforman la molécula de paracetamol cuando se ordenan de la manera adecuada. La química médica, aquella que se dedica a encontrar nuevos fármacos para diferentes enfermedades, busca en esencia cómo combinar estos átomos para encontrar nuevos fármacos. ¿Por qué es tan difícil? Bueno, si uno busca todas las combinaciones posibles de átomos que forman potenciales fármacos, obtiene la friolera cantidad de 1060 candidatos. Teniendo en cuenta que el Sol está compuesto por alrededor de 1056 átomos, os podéis imaginar el tamaño del pajar donde está escondida esta aguja. Sintetizar, almacenar, y evaluar esa cantidad de moléculas es, lógicamente, inviable. Menos mal que la quimioinformática está de nuestro lado.
Buscando llaves para cerraduras imposibles
La quimioinformática es un concepto general que engloba el uso de la informática y la computación para solucionar problemas relacionados con el desarrollo farmacéutico. El motivo está claro: los ordenadores son máquinas extremadamente eficientes de hacer cálculos. En apenas una fracción de segundo, son capaces de resolver operaciones matemáticas que a cualquier persona le supondría toda una vida y un gasto inviable en analgésicos. Afortunadamente, el efecto terapéutico de un fármaco puede transformarse en modelos matemáticos.
Vamos a plantearlo de la manera más sencilla posible: supongamos que, como sucede en muchas enfermedades, existen proteínas aberrantes que, debido a su mal funcionamiento, contribuyen al origen y progreso de una patología. La misión de los fármacos es interaccionar con estas proteínas, impedir su mal funcionamiento, y corregir la enfermedad. Por ejemplo, el ibuprofeno es el encargado de unirse a la proteína COX-2, impidiendo que esta produzca una serie de efectos proinflamatorios. Si COX-2 fuera una cerradura, el ibuprofeno sería una llave que encaja con precisión nanométrica. Esto es lo que está detrás de su efecto analgésico, ni más ni menos. Si conocemos la proteína responsable de la enfermedad que queremos combatir, basta con encontrar la molécula apropiada. ¿Cómo exploramos ese inmenso espacio químico? ¿Cómo encontramos la molécula ideal dentro de esos 1060 candidatos?
Un recurso ampliamente utilizado son los cribados virtuales. Los ordenadores son capaces de, en primer lugar, modelar una estructura tridimensional de nuestra proteína de interés. En otras palabras, es capaz de aportarnos una imagen de su estructura, cómo funcionan sus componentes, y averiguar si dispone de alguna región adecuada donde puedan unirse moléculas de tipo fármaco. Una vez identificada esta cerradura, los ordenadores criban de manera virtual cientos de miles de moléculas en un intervalo de tiempo razonable. Todo esto lo realiza con un algoritmo de puntuación complejo donde se calcula la complementareidad electrostática (cargas que se atraen o se repelen) y estérica (cómo de bien encajan los volúmenes de la proteína y la molécula). En román paladino, aquellas moléculas que encajen mejor con la cerradura obtienen una puntuación más alta.
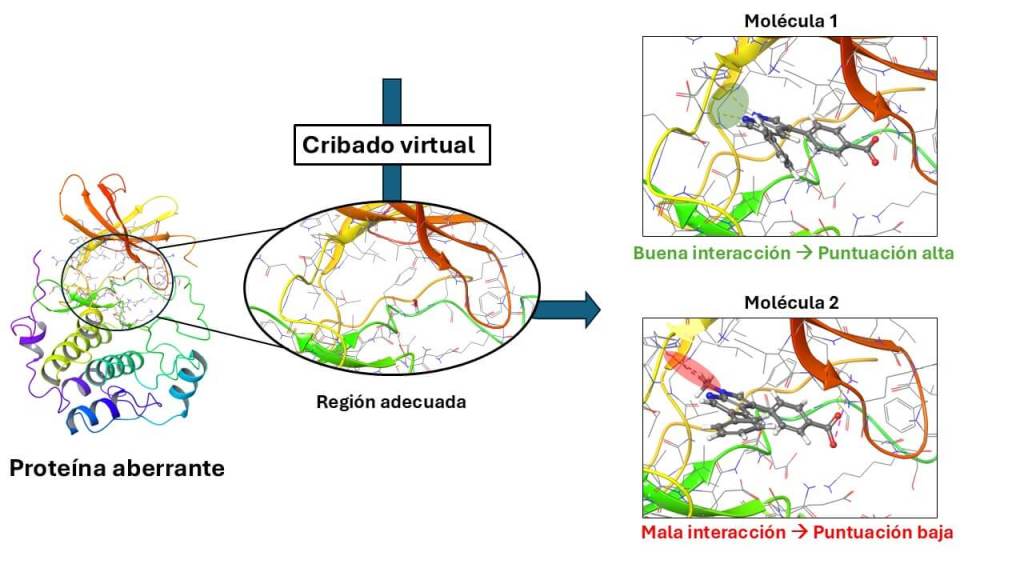
Esto permite descartar de un plumazo una inmensa cantidad de moléculas inactivas que, según el algoritmo, obtienen una baja puntuación, quedándonos con solo unas pocas decenas de estas. Desde luego, un número bastante más manejable que del que partíamos. Si la suerte te pilla trabajando, es muy probable que dentro de esas decenas de moléculas haya un potencial fármaco. Y esto no termina aquí. Si conseguimos un candidato, siempre podemos recurrir de nuevo a los ordenadores para estudiar cómo se une a la proteína. Esto nos permite proponer nuevas modificaciones, nuevos grupos de átomos que añadirle a nuestra molécula para convertirla en una verdadera competidora en la carrera contra la enfermedad que estamos combatiendo.
Resolviendo rompecabezas con la quimioinformática
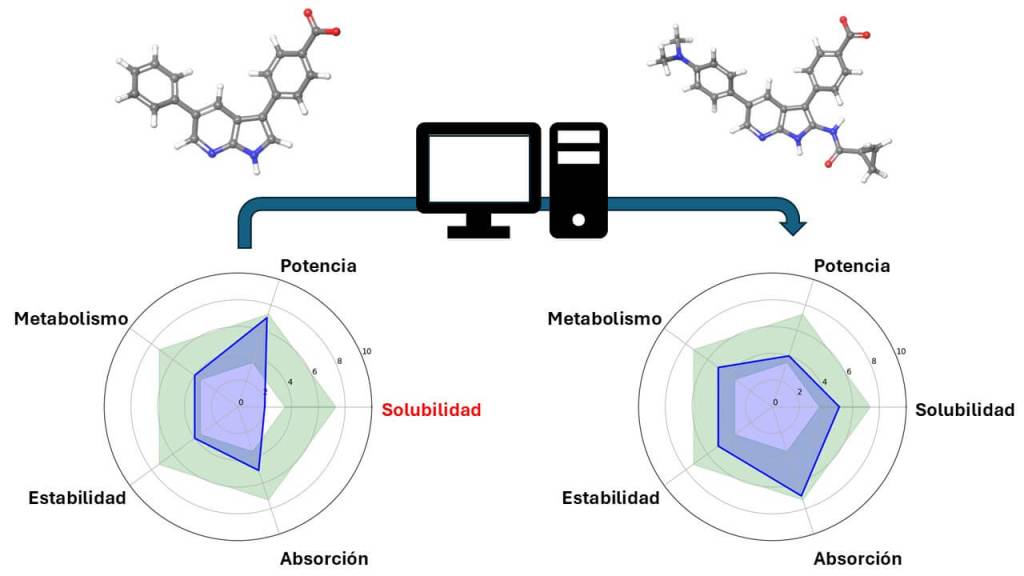
Los que nos dedicamos a la química médica somos conscientes del colosal proyecto que supone empujar una molécula desde cero al mercado. De media, el proceso suele durar entre 10 y 15 años. Es un cubo de Rubik algo extraño. El que todos conocemos tiene 6 lados y puede resolverse con un mínimo de 20 movimientos. El del descubrimiento de fármacos es un cubo con al menos 30 caras. La potencia, solubilidad, absorción, metabolismo, estabilidad, y un largo etcétera de propiedades, conforman este hercúleo rompecabezas. Cada vez que mueves una cara, afecta al resto del cubo. Sin embargo, contamos con una ventaja: no es necesario que todas las caras estén perfectamente alineadas para que el cubo funcione. No se necesita el fármaco más potente, se necesita que sea lo bastante potente para ejercer su acción terapéutica. De igual manera, no se necesita el fármaco con mayores valores de absorción en el organismo. Más bien, necesitamos que se absorba lo suficiente para ejercer su acción terapéutica. Todas estas caras del cubo, incompletas por sí solas, se compensan unas a otras, permitiendo que nuestra molécula sea la candidata perfecta. Y como antes, es la combinación de átomos que componen nuestra molécula la que determina todas estas propiedades.
En quimioinformática, disponemos de algoritmos que intentan predecir todas estas variables. Supongamos que tenemos un conjunto de moléculas con una propiedad interesante. Por ejemplo, moléculas extraordinariamente solubles en agua, las cuales pueden encontrarse en la naturaleza o que han sido descubiertas a lo largo de décadas de investigación. Si nuestro candidato a fármaco presenta problemas de solubilidad, la inteligencia artificial es capaz de inspeccionar estas moléculas, analizar qué combinación de átomos son los responsables de esta propiedad, y proponer nuevas modificaciones en nuestro candidato a fármaco. Y lo que es más importante, podemos predecir no solo cómo se ha visto mejorada su solubilidad, sino el impacto de esta modificación en el resto de sus propiedades. Si se mantienen en unos márgenes adecuados, el cubo de Rubik estará más cerca de completarse. Tras varios ciclos y toneladas de trabajo experimental que validen las predicciones, nuestro candidato a fármaco se encontrará más cerca de ser dispensado en las oficinas de farmacia.
Si algo ha quedado demostrado es que la quimioinformática ha transformado la manera en la que diseñamos fármacos. Supone una reducción de costes y tiempo innegable, y es capaz de proponer soluciones que, siempre supeditadas al criterio de los químicos médicos, acelera el desarrollo de nuevas y mejores moléculas. Y por si no fuera suficiente, permite gestionar el vértigo que produce pensar en el universo químico que hay dentro de un comprimido farmacéutico. Para nosotros, esto ya es motivo suficiente.
Referencias
- Tran-Nguyen VK, Junaid M, Simeon S, Ballester PJ. A practical guide to machine-learning scoring for structure-based virtual screening. Nat Protoc, 16 de octubre de 2023. DOI: 10.1038/s41596-023-00885-w
- Paul D, Sanap G, Shenoy S, Kalyane D, Kalia K, Tekade RK. Artificial intelligence in drug discovery and development. Drug Discov Today, 20 de enero de 2025. DOI: 10.1016/j.drudis.2020.10.010

Enrique Madruga Mayordomo
Graduado en Farmacia por la Universidad de Salamanca. Máster en Descubrimiento de Fármacos por la Universidad Complutense de Madrid

Cortesía de Muy Interesante
Dejanos un comentario: